Информация носит справочный характер. Не занимайтесь самодиагностикой и самолечением. Обращайтесь ко врачу
.
 Болезни, обусловленные мутациями в митохондриальной ДНК, проявляются не при рождении, а во второй, реже – в первой декаде жизни.
Болезни, обусловленные мутациями в митохондриальной ДНК, проявляются не при рождении, а во второй, реже – в первой декаде жизни.
В ряду генетических заболеваний числится Синдром Кернса–Сейра (Kearns–Sayre syndrome, KSS) — митохондриальная миопатия, поражающая глаза и мышцы пациентов. Неоднозначная динамика симптомов, их не одновременное проявление, а также отказ от биопсии и детальных генетических исследований осложняет точную постановку диагноза.
История появления диагноза
В 60-е годы доктора медицины Томас Кернс и Джозеф Сейр обратили внимание на три симптома, встречающиеся одновременно (нарушения зрения, пигментацию сетчатки глаза, нарушения проводимости нервных импульсов в сердце), описали несколько клинических случаев протекания болезни. Только в конце 80-х годов, после проведения исследований в области изменений в митохондриальных структурах стало понятно, как связанны между собой различные нарушения здоровья у одного пациента.
Синдром получил название по именам специалистов, впервые описавших характерную клиническую картину.
Дальнейшее изучение KSS показало, что симптоматика синдрома может быть более вариабельной (мышечная недостаточность в конечностях, слабоумие, нарушения в других сенсорных системах, общая задержка развития, низкий рост), но речь все равно идет о системном заболевании из-за нарушений в ДНК митохондрий.

Мнение врача:
Синдром Кернса–Сейра – это редкое генетическое заболевание, которое характеризуется нарушением функции нервной системы и мышц. Врачи отмечают, что симптомы этого синдрома могут включать в себя мышечную слабость, нарушения координации движений, проблемы с речью и затруднения в дыхании. Диагностика основывается на клинических проявлениях и генетических исследованиях. Лечение синдрома Кернса–Сейра направлено на симптоматическую терапию, включая физиотерапию, лечебную массаж и речевую терапию. Важно подчеркнуть, что раннее выявление и комплексный подход к лечению могут значительно улучшить качество жизни пациентов с этим редким заболеванием.

Патогенез и общий характер симптоматики
Среди зафиксированных случаев (всего около 300 детально описанных), только в нескольких можно говорить о семейной наследственности (у родителей зафиксированы те же симптомы, что и у детей). Остальные — мутации на ранних сроках беременности, подлежащие диагностированию после 4 года жизни ребенка.
Симптомы при синдроме Кернса Сейра развиваются по нарастающей:
- уменьшение двигательной активности;

- значительное ухудшение четкости зрения;
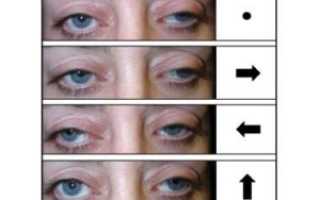
- птоз одного или двух век;
- минимизация движений глазным яблоком до полного их отсутствия;
- брадикардия, аритмия;
- нарушения слуха, координации движений;
- проблемы с глотанием пищи и слюны (реже);
- атрофия мышц конечностей;
- прекращение работы одного из желудочков сердца и смерть.

Этиология болезни такова: «испорченный» геном митохондрий продолжает копироваться в искаженной форме при каждом делении клетки. Поскольку среди его главных функций – нормальный энергетический обмен, то начинают страдать те органы, где затраты энергии максимальны (нервные волокна, сенсорная система, мышцы). При отсутствии симптоматического лечения митохондриальной миопатии наступает смерть (средние показатели — до 24-36 лет).
-
Читайте также:

Примечательно, что параллельно с «испорченными» клетками развиваются и работают те, у которых митохондрии в норме.
Генетический аспект
Митохондриальные проблемы передаются по материнской линии при значительном количестве измененных генов в органелле. На практике это означает, что у здоровой по виду женщины-носителя могут родиться и здоровые, и больные дети.
Наука пока не выявила точной закономерности развития синдрома. Понятно, что важным является первый триместр беременности, когда происходит активное деление клеток и закладка систем органов. Какой именно фактор влияет на более активное деление дефектных клеток, чем здоровых, пока не выявлено.
Синдром Кернса–Сейра встречается одинаково часто у мужчин и женщин разных рас.

Опыт других людей
Синдром Кернса-Сейра – редкое генетическое заболевание, вызванное мутацией гена KRT5 или KRT14, приводящей к характерным симптомам, таким как образование пузырей на коже и слизистых оболочках. Диагностика основывается на клинических признаках и генетических тестах. Лечение направлено на симптоматическую терапию, включая уход за кожей, применение мягких повязок и предотвращение инфекций. Люди, столкнувшиеся с этим синдромом, отмечают его сложность, но подчеркивают важность поддержки со стороны близких и специалистов для улучшения качества жизни.
Характерный клинический случай
Синдром Кернса–Сейра может иметь разные первоначальные проявления и дальнейшую клиническую картину. К примеру, у 25-летнего мужчины с выраженным птозом обеих век, атрофией мышц конечностей и спины, отсутствием движений глазными яблоками, пониженным зрением, болезнь проявилась в младшем школьном возрасте.
При физических нагрузках (бег) появлялись сильные головокружения, рвота, боль в животе. В 12 лет специалисты заметили птоз левого века, через 5 лет такая же участь постигла правое веко. При этом отмечалось двоение в глазах.
На фоне симптоматического лечения опущение уменьшилось, но препараты помогали все меньше, что сочли последствием привыкания. Для предотвращения полной слепоты рекомендовали укорочение век. С 23-летнего возраста начала развиваться тугоухость, прогрессирует общая мышечная слабость.
Нарушений в интеллектуальном развитии, работе сердца не обнаружено. Постоянное наблюдение у кардиолога и невропатолога дает возможность стабилизировать состояние, снизить интенсивность прогрессирования болезни.
-
Читайте также:


Симптомы, характерные для синдрома Кернса–Сейра
Митохондриальная миопатия может развиваться и другим путем, когда при раннем выявлении болезни прослеживается быстрое ухудшение состояния без отзыва организма на лечение. Например, родители 5-летнего мальчика обратились к врачам с жалобами на одновременный птоз обеих век, нарушения движений глазными яблоками. При дальнейшем наблюдении выявили также нарушения сердечного ритма, психофизическое отставание в развитии, торможение полового созревания. За 6 лет наблюдений синдром прогрессировал до:
- координационных нарушений;
- затрудненного жевания и постоянного поперхивания;
- атрофии мимических мышц;
- атриовентрикулярной блокады;
- наджелудочковых экстрасистол.
Главным достижением лечения в данном случае стала временная нормализация сердечного ритма.
Динамика синдрома индивидуальна, зависит от количественных изменений в митохондриях, систематически принимаемых препаратов, а также от сопутствующих болезней.

Диагностический подход
Точное диагностирование заболевания возможно только при проведении биопсии мышц на предмет выявления патологии. Косвенными показателями болезни являются:
- совокупность симптомов и их динамика;
- завышенные уровни пировиноградной и молочной кислот в крови пациента;
- отсутствие отклика на прозерин, калимин;
- данные электрокардиографии;
- исследования на возможные нарушения в работе эндокринной системы (выявлены у 3% пациентов, существенно усугубляют протекание болезни).
Выявление болезни обычно занимает длительный промежуток времени ввиду медленного нарастания симптомов, неготовности родителей к проведению биопсии и детального генетического анализа.
Возможности современной медицины

Оперативное подрезание век
-
Читайте также:

Синдром Кернса Сейра не поддается полному лечению на данном этапе развития медицины. Предполагается, что возможно купирование всех симптомов при стимуляции к делению здоровых клеток, постепенному замещению ими больных волокон.
Механизм подобной операции не разработан.
Экспериментальные операции показали, что спутниковые клетки могут способствовать регенерации мышечных волокон.
Пациент может рассчитывать на:
- вживление кардиостимулятора для предотвращения остановки сердца;
- введение коферментов для понижения уровня пировиноградной и молочной кислот (наблюдается улучшение подвижности глаза);
- укорочение век (временно улучшает зрение);
- использование слухового аппарата;
- нормализацию уровня гормонов.
Все названые меры снижают дискомфорт в быту, но не предотвращают полностью дальнейшее развитие мышечной слабости.
Последствия и прогноз
Митохондриальная миопатия имеет не оптимистический прогноз: прогресс мышечной слабости до остановки сердца или дыхания. При надлежащем лечении возможно существенное увеличение продолжительности жизни, но с признанной инвалидностью (2-3 группа).
В любом случае, речь идет о пожизненном наблюдении у кардиолога и невропатолога с постоянной корректировкой препаратов, их доз в зависимости от показателей анализов крови.
Профилактических методов нет, ввиду незначительной изученности пусковых механизмов.
Действенным методом уменьшения негативного влияния синдрома на качество жизни станет своевременное обращение к врачу, прохождение всего комплекса обследований и начало симптоматического лечения.

Перед запланированной беременностью родителям стоит пройти тест на определение возможных генетических отклонений у общего ребенка.
Частые вопросы
Что такое синдром Кернса–Сейра?
Синдром Кернса–Сейра — редкое наследственное заболевание, характеризующееся нарушением митохондриальной функции. Генетические мутации в генах, кодирующих митохондриальные белки, приводят к дефектам в производстве клеточной энергии.
Каковы основные симптомы синдрома Кернса–Сейра?
Наиболее частые симптомы включают прогрессирующую мышечную слабость и истощение, затруднения речи, дисфагию (нарушение глотания), внешнюю офтальмоплегию (паралич глазных мышц), а также соматические проблемы, такие как сердечная недостаточность и нарушения дыхания.
Полезные советы
СОВЕТ №1
При подозрении на синдром Кернса–Сейра обратитесь к генетику для проведения соответствующих генетических тестов для точного диагноза.
СОВЕТ №2
Регулярно наблюдайтесь у врачей различных специальностей (невролог, офтальмолог, кардиолог и др.) для контроля состояния здоровья и своевременного выявления осложнений.
СОВЕТ №3
Обсудите с врачом возможные методы лечения синдрома Кернса–Сейра, включая симптоматическую терапию и реабилитационные мероприятия.